
Gordon Dougan & Kat Holt
Making a COVID-19 vaccine will be a huge undertaking. We have outlined here, in some detail, the steps normally taken to make a vaccine and compare this to what may happen for COVID-19. It will be a race full of challenges and surprises so hold your breath.
(Spanish translation available here)
Introduction
Making a new vaccine from scratch is a technologically challenging process. There are normally many steps needed to achieve this and as each step is taken, the cost increases by at least an order of magnitude and hence the risk. The development of a new vaccine can fail at any time. Here is an outline of the traditional steps taken. We will then describe how the process may be shortened for COVID-19 vaccines.
First up, what is in a vaccine?
A vaccine is a ‘medicine’ that is used to protect people and animals against a specific infectious disease e.g. polio or mumps (1, 2). Vaccines work by inducing our bodies to generate ‘protective immunity’, in a similar manner to when we are infected with a virus or bacteria, but without actually causing disease. They need to be as safe as possible as they actually prevent (they are prophylactic) rather than treat diseases and are often used for children.
There are a number of different types of vaccines so we need to think about which type might work for COVID-19. The goal is to make something we call an antigen that looks as close as possible to the virus, but cannot actually cause disease. We can then safely present this to the human immune system as a vaccine, to train the system to recognise and respond to the actual virus when it encounters it for real (3).
Classical vaccine types are:
- Live attenuated vaccine. This is based on a form of the virus/bacteria that can still grow to a limited extent inside humans, and thus immunise and protect, but has been attenuated/modified so that it can no longer cause disease.
- Inactivated or killed/dead vaccine. This is normally based on the whole virus/bacteria or a simple protein (or for bacteria sometimes carbohydrate) component. The whole virus, protein or parts of these can be used for the vaccine and are known as the antigen.
- Vectored vaccine. A live or semi-replicating vaccine based on a hybrid between the virus/bacterial vaccine target antigen and another virus that acts as a delivery system.
- A DNA or RNA vaccine. This is based on the genetic material of the target virus/bacteria rather than the actual virus particle itself. Once injected or delivered the genetic material expresses the protective antigen.
There are other types of vaccines and they can sometimes be used in combination in so called prime/boost strategies but we will not consider these here. Before looking at each of the above vaccine options in terms of a COVID-19 vaccine, let’s have a look at the overall vaccine development process.
An overview of the whole vaccine development process
Project establishment
Vaccine development is complicated. Thus, an early step in vaccine development (normally just after the Discovery phase, see below) is to form a project management team who have the job of delivering the vaccine and planning a clear and considered path that eventually leads to the licensing and use of a vaccine (4). Vaccine development is normally a formally managed process using milestones and targeted deliverables that are set right throughout the project. The project team take the longer view covering the whole process.
The project management team will undertake costings, preclinical planning and arrange each of the steps we outline below. They will also begin a dialogue with the regulators or regulatory authorities who will eventually decide if the vaccine is fit to be licensed. The team also tries to identify any potential road blocks to the process and eventually will hand over to other teams such as manufacturing, clinical trials and marketing.
Getting a licence and selling vaccines
Figure 1 shows the classical path to vaccine licensing (5). This is a standard model but it has flexibility.

The road to actually licensing and selling a vaccine is incredibly expensive and full of perils. Most vaccine discovery projects make good press releases but very few make it through this path. Each country has its own path to licensing a vaccine. The decision to license is usually taken by a regulatory authority under the control of a government (6). For the USA this is the Food and Drug Administration (FDA) (7). For the European Union it is the European Medicines Agency (EMA) (8), which recently moved from London to Amsterdam.
A dossier for each vaccine is built up and a dialogue begins between the manufacturer and the regulators. This is balanced but is a mechanism of working the process to a positive end: a safe, effective and manufacturable vaccine.
Unfortunately, many countries do not have a functional National Regulatory Authority (NRA) that can approve their medicines effectively. This is a sad truth in the modern world. This means that if a vaccine is made in a country without a recognised NRA it is difficult or impossible to sell the vaccine into other countries or markets. Also such countries can be the victims of unregulated or even fake medicines.
The WHO has a system of inspection and validation that can apply a standard of approval called WHO Prequalification (9). If a vaccine or a vaccine manufacturer can gain WHO Prequalification of a vaccine it helps sell the vaccine more broadly. It is a signature of quality. WHO Prequalification is tied up with effective NRAs so the situation can get complicated quickly (for another day!).
If a licence is granted then the developers can breathe a sigh of relief but the process is not completely over yet (10). Once a vaccine is approved, further post licensure studies are carried out to look for any safety signals, efficacy problems or vaccine batch issues. It can also be useful to see how the vaccine works in different age groups or in different social or geographical settings.
Making COVID-19 vaccines
Discovery Phase
The first phase of making a vaccine is known as the Discovery Phase. This is all carried out in laboratories. It can start in a normal academic laboratory but the project then has to move to an approved location for making vaccines, a so-called Good Laboratory Practice or GLP lab where everything is documented including people, processes, equipment and reagents (11).
To simplify approval it is usually best to start with materials, including antigen expression and delivery systems (12), which have been validated previously for making vaccines or other medicines. This may not be completely true in the rush to make a COVID-19 vaccine.
Remember the goal here is to make a vaccine that looks like SARS-CoV-2, the virus that causes the disease COVID-19, but cannot itself make people sick.
(a) Live attenuated vaccine
People are considering all approaches but a live vaccine presents some challenges for COVID-19 in terms of safety and stability. Some countries might go for it. However, growing the virus itself is difficult so let’s leave this option for now.
(b) Inactivated or dead vaccine
This is an option. We would not use the whole SARS-CoV-2 virus particle but would use a protein component. At the moment the favoured antigen is the virus spike protein (13) or possibly the actual tip of the spike protein known as the tip domain. This is the part of the virus that it uses to attach itself to human cells. It is also a weak point of the virus as it is the part that antibodies (and immune cells) target to stop or neutralise the virus (Figure 2) (14).
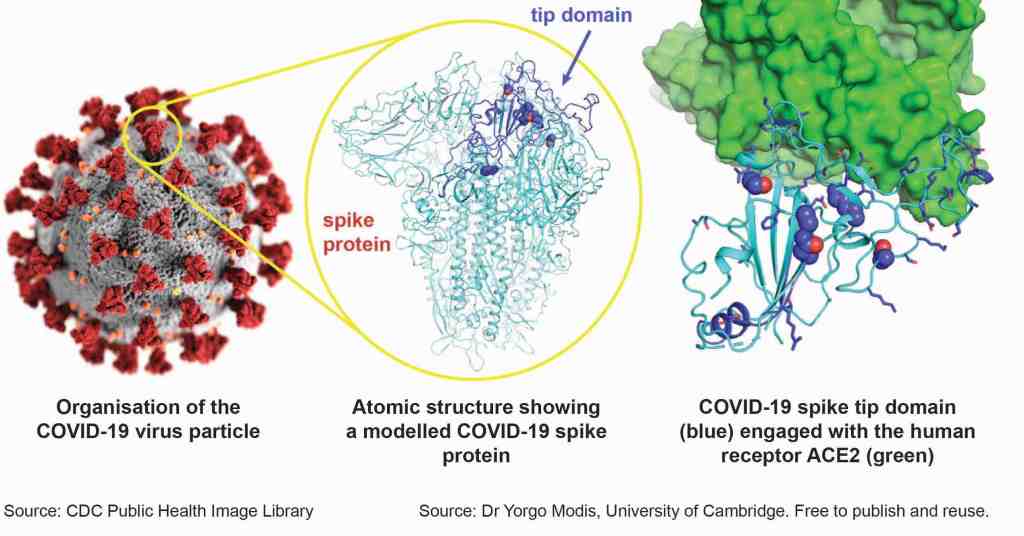
Other SARS-CoV-2 proteins may be considered as vaccine components as they may be antigenically conserved or are targets of cellular immunity.
Making the protein
For vaccine development, the virus protective antigen is normally expressed as a recombinant or cloned protein in some kind of protein production platform that can be used at scale in industry (15). The host is normally a bacterial, yeast or eukaryotic tissue culture cell (e.g. mammalian or insect cell). Essentially, the gene coding for the viral protective antigen is moved from the virus into a heterologous host organism that then produces the protein.
The protective antigen has to be properly structurally folded so that it looks exactly (or almost exactly) as it does on the actual virus. Sometimes stabilising mutations have to be introduced into the protein to increase expression or shelf life of the antigen. SARS-CoV-2-like spike and tip protein domains do not easily fold properly in bacteria, so yeast (e.g. Saccharomyces cerevisiae or Pichia pastoris) will likely be used (see ref 16, as example of a company using Pichia technology).
Alternative host cells can be insect (e.g. for Baculovirus) or mammalian cells grown in culture but this may be expensive. Yeast can be grown in huge fermenters, are already used in the pharmaceutical industry to make vaccines and can produce kilograms of protein. This is the likely way forward, although posttranslational issues such as glycosylation will need to be managed.
Once the protein is made it has to be carefully assessed for stability and tested to see if it can be recognised by neutralising antibody or some other protective immune mechanism.
Next step, formulation begins
The antigen will be assessed for any potential toxicity to make sure it does not do anything inappropriate biologically (e.g. is toxic or stimulates the wrong cells). If this is the case, it can be further inactivated using chemicals or by genetic engineering.
The protein on its own may not be powerful or immunogenic enough to stimulate protective immunity. It may need an adjuvant (an immunostimulator) to work properly. There are only a few licensed adjuvants that can be used in humans and most of these, with the exception of an old adjuvant Alum (a form of aluminium salts) are owned by companies. This may be a bottleneck for this approach.
Modern licensed adjuvants include MF59 and some members of the AS01 series produced by GSK (17). Antigens are usually tested with different adjuvants in animals to choose the right one, although animals are not always a good predictor of human responses. This is part of so called preclinical evaluation. The vaccine may be further formulated to improve shelf life (stability on storage) etc.

Concerns
There are some concerns about using a protein-based (and indeed other) vaccine for coronaviruses. Work in the 1960s on another virus, Respiratory Syncytial Virus (RSV), which causes lung infections in children, showed that a killed version of this virus can induce vaccine enhanced disease and this stopped the vaccine development. Vaccine enhanced disease can theoretically occur just after vaccination or when a vaccinated person encounters the real virus. Essentially, it is an immune pathology or dysfunction. It is a rare event and is normally identified in the vaccine development path, avoiding damage after a licence is granted.
Similar concerns have been identified for experimental coronavirus vaccines based on proteins (18), but these have only so far been seen in animals. Therefore, care will be needed if this type of vaccine is developed. Here, the choice of adjuvant might be important to direct the immune response in a safe direction.
(c) Vectored vaccines
A vectored vaccine can be used to deliver an antigen. Vectored vaccines are considered if (i) the original virus/bacteria is too fastidious (hard) or dangerous to grow, or (ii) if the original virus/bacteria is antigenically very complex and you want to simplify the components of the vaccine. Vectors have to be safe in themselves, so they are usually attenuated or disabled genetically so they immunise without causing disease.
Vectored vaccines have their own adjuvant activity and so do not normally need an adjuvant. Vectors can be bacteria or viruses. As SARS-CoV-2 proteins likely do not fold in bacteria, let’s consider viral vectors.
There are several viral vector systems based on viruses such as adenoviruses, vaccinia pox viruses and vesicular stomatitis virus (VSV). Let’s focus on adenoviruses but we do not rule out the other options (19).
Adenoviruses are DNA viruses that can infect multiple cell types and are used both in vaccines and for gene therapy (e.g. against cancer or to correct genetic defects). They have relatively small genomes and can be easily genetically modified to make them ‘safe’ for human use. There are many types of Adenoviruses and vectors generated from them.
When used as a vector some of the proteins of the adenovirus are replaced by the gene for the antigen of the vaccine target. For COVID-19 this would likely be the SARS-CoV-2 spike protein or the tip protein (see Figure 2), but could be other proteins. Figure 4 shows the principle of how the vaccine vector is constructed.
Once constructed the adenovirus-vectored COVID-19 vaccine would be tested for stability and preclinically in cellular, animal and other immunogenicity studies. The aim here is to see if the vaccine can induce protective immunity, perhaps in the form of neutralising antibodies (20).
We have comparatively limited experience in humans of using adenovirus-vectored vaccines but the approach is rational and certain teams have built up experience with these systems (e.g. with Ebola vaccines).
(d) RNA or self-replicating RNA vaccines
These are the very newest form of vaccines that are about to be road tested with COVID-19. RNA is the intermediate between DNA and protein in the so called central dogma of molecular biology (21). If RNA can be placed properly into cells, it can direct production of the protein it codes for by harnessing the cells’ own protein synthesis machinery.
RNA vaccines are based on synthetic RNA that is chemically synthesised. This synthetic RNA is based on the genetic code of a backbone virus which is not SARS-CoV2, rather like the vectored vaccines. RNA viral systems can be based on a-viruses such as Semliki Forest Virus that can direct the target cell to express the viral protective antigen (22).
The gene coding for the SARS-CoV-2 antigen (e.g. spike or spike tip protein) is synthesised and placed into the synthetic RNA, at a point where the host cell will be directed to make the protein antigen (Figure 5).
RNA itself cannot easily get into cells so the synthetic RNA has to be formulated into a particle that behaves like a virus but is safe. These so called packaging and delivery particles can be based on fatty or lipid structures known as liposomes. These liposomes can be formulated to improve stability, efficiency of delivery and other factors.
Some RNA vaccines are so-called self-amplifying in that once the synthetic RNA is inside the cell it can make more and more copies of the part of the RNA encoding the protective antigen (23). This can amplify the protective signal.
RNA based vaccines are still relatively untested and it will be interesting to see how they perform in real trials. It is noteworthy that DNA-based vaccines have been tested in humans but so far, they have fallen short in terms of immunogenicity.
Further phases of vaccine development
Preclinical phase
The preclinical phase covers all of the work required to get a vaccine ready for clinical trials in humans. The Table below lists some of the activities that would go on traditionally in this phase to satisfy both regulatory and ethical standards as defined in the particular country where the development is being undertaken.
The aim of the preclinical phase is to do as much testing as possible on toxicology, immunity, formulation, animal testing, identifying correlates of protection, assay development and the scaling up of antigen production. Lots of troubleshooting and fine tuning.A key aim is to make a batch of material under Good Manufacturing Practice (GMP) conditions (24). This means that the vaccine is made under controlled, auditable and safe conditions ready for use in humans. Many countries have a strict requirement for a GMP product if it is to be used in humans. GMP production is expensive and can cost $1M or more. GMP requires special facilities and expertise.
| Pre-Clinical Development |
| Vaccine construct design |
| Upstream process development |
| Formulation |
| Analytical development |
| Potency assay development |
| Product characterization studies |
| Animal immunogenicity |
| Technology transfer to a GMP Contract Manufacturing Organization (CMO) |
| Engineering batches by CMO |
| GMP cell banking |
| Toxicology studies |
| Stability data |
| Clinical Development |
| Dossier submission for Phase 1 IND application |
| Regulatory approval for Phase 1 |
| Phase 1 clinical trial |
Table 1: Key tasks undertaken during pre-clinical and clinical vaccine development.
Source: Sourabh Sobti, Hilleman Laboratories (25).
Phase 1, into the clinic
The Phase 1 study is usually the first assessment of a vaccine in humans. It is normally performed under strict ethical and regulatory conditions and is in the hands of clinical specialists. The trial is there to assess safety as a first priority and everything is normally designed around this consideration. It is also the first opportunity to assess aspects of the immune response to a vaccine in humans.
Such studies are normally performed in healthy adult volunteers from the country where the vaccine is being developed. Some initial immunological data can be collected and dosing strategy can also be evaluated. There is usually a control arm where people are given a placebo. This might be a related vaccine or just the same adjuvant as used in the vaccine.
The numbers in Phase 1 studies is normally quite small and the vaccination team are normally blinded in that they do not know which individuals are receiving vaccine or placebo (26). The rules for the design of Phase 1 studies are not set in concrete and different investigators, companies or countries will set these up as they see fit. Also, there will be different requirements for different vaccines.
Some people will be excluded from Phase 1 and later studies such as those already exposed to the virus/bacteria under study or individuals with medical complications such as being immunocompromised. Special checks are made to pre-screen people.
If a Phase 1 trial is performed in a particular setting such as a richer country, then it might be repeated elsewhere, for example in a poorer setting where the disease might be common. For example a vaccine on cholera might be trialled in this way.
Phase 2
This is normally the next step in the development of a vaccine and it has somewhat more flexibility than Phase 1. The numbers of people enrolled is normally larger than Phase 1 and participants may not be healthy adult volunteers if the ultimate target population is a specific social group such as children or the elderly. Sometimes special groups such as pregnant women or those already infected (for a therapeutic vaccine) might be considered.
In the Phase 2 studies more complex immunological, physiological or biochemical data might be collected and attempts made to identify correlates of immunity e.g. neutralising antibodies, that might help in the licensing of the vaccine (27).
If a Phase 2 trial is performed in an area where the target disease is endemic or prevalent then some efficacy or information on the ability of the vaccine to protect may be collected. Indeed Phase 2 trials are usually performed where the vaccine might be used, although this is not always the case. Indeed, where trials are conducted is critical for many reasons including licencing. Also, trial should be conducted where possible using people who will have access to the vaccine once licenced.
In recent years the use of so called Controlled Human Infection Models or CHIMS have returned as methods for quickly obtaining data on protection (28). In a CHIM human volunteers are vaccinated (or given a placebo if they are controls) and then infected/challenged with the actual virus/bacteria or an attenuated form of it. CHIMs need incredibly careful ethical and clinical management but they can shortcut the time to licensing a vaccine. However, regulators and policy makers are now more likely to take them into consideration for licencing purposes.
Phase 3
Phase 3 studies are the classical way to evaluate the efficacy or protective power of a vaccine (29). These can involve thousands of people and be very expensive. They are normally carried out in the field e.g. in the places where the disease is present or the people to be vaccinated can be found. i.e. part of a city or perhaps a healthcare setting such as a group of hospitals.
The design and control of Phase 3 studies is complex and challenging. It requires careful consideration of many factors including:
- The nature of the control groups (those not receiving vaccine but placebo or equivalent).
- The social and geographical lay out of the vaccine field site.
- The ethics of the trial, particularly in a health emergency (30). Specialised impartial groups are set up to oversee trials to prevent bias. These groups have the power to stop trials for several reasons such as adverse safety signals or early proof of protection.
- The statistical power of the trial and the clinical endpoints (31). If diseases are rare then very large numbers may be required to demonstrate statistically significant protection. Also, for some vaccine classes, a correlate of immunity might be measured in place of actual clinical parameters or disease. This is largely a regulatory consideration/call.
- There are certain ethical and logistical challenges if you are trying to replace an existing vaccine with a new better vaccine. There is usually a need to test equivalence (note, this is not an issue with COVID-19).
Regulatory and license to sell
The ultimate aim of making a vaccine is to get it to those who need it, not as simple as it sounds.
National and International regulatory bodies were established to enforce quality and high standards on medicines. They require proof that a vaccine is both effective and safe. The potential manufacturer or seller has the responsibility of delivering this proof as a regulatory dossier or equivalent.
Each regulatory authority has its own processes. Regulators can help with fast tracking but they will try to stick to established procedures. This is not always possible in a crisis with significant political pressures of course. Also, the ‘label’ on the product is surprisingly important in that it designates where, how and on whom it can be used. More about this another time.
Post licensure
After licensing the vaccine has to be manufactured at scale under GMP to meet the demand (32). This sounds easy. It is NOT. Manufacturers may have to quickly build state-of-the-art facilities at some risk. These will be heavily inspected to ensure the quality of the manufacturing process. This is a whole science in itself.
Maintaining manufacturing facilities and filling them with highly trained, process-compliant staff is also not easy. Many countries do not have the infrastructure to support this. Indeed, most countries do not.
Further studies on the performance of the vaccine in terms of safety, different populations and efficacy continue following licensing in Phase 4 trials. Also it is vitally important to know the duration of and type of immunity a vaccine induces. This may vary in different locations due to factors such as endemic disease exposure and environment.
Some vaccines are used routinely (e.g. vaccines in the Expanded Programme on Immunisation (EPI) (33), some only in certain regions (e.g. typhoid vaccines). Some are stockpiled for use in an outbreak or emergency (e.g. cholera vaccines) (34).
What about a COVID-19 vaccine?
Much of the above will go out of the window in an emergence such as COVID-19 (see Figure 1). Governments have the power in most countries to completely overrule the regulatory authorities. They can gamble and take short cuts, based on a risk assessment of sorts that takes into account the nature of the emergency. The EMA provides clear guidance on how vaccines might be fast-tracked in emergencies (8).
Most of the preclinical phase can be dispensed with (a choice) and the minimal work can be undertaken, even with unproven delivery platforms so pressure builds up on everyone. Decisions have to be taken in real time, clever clinical trial designs e.g. adaptive designs might be used.
Theoretically, a human challenge could be used to validate a COVID-19 vaccine but there are enormous ethical considerations here (35). Most challenge studies require a validated treatment such as an antibiotic (for bacteria) and an anti-viral (for a virus) so that volunteers have an acceptable level of protection from developing serious disease. Hopefully these will come through for COVID-19.
It would be challenging to set up Phase 3 efficacy studies for COVID-19 under present conditions. However some form of hybrid or adaptive trial is more likely based on safety, immunogenicity and indirect protection estimates. This is hard to call at the time of writing.
Some developers may try to use an immunological correlate of protection to license a COVID-19 vaccine. This could be a neutralising (blocking) antibody titre that prevents viral growth or even kills the virus. The problem is that for many vaccines in vitro neutralising titres do not correlate with protection e.g. rotavirus vaccines. Some viruses need intracellular as well as extracellular neutralisation, complicating the situation. Thus, efficacy studies, before or after licensing, may still be required if no proven correlate is identified.
Some countries have agencies that establish standards for testing, correlates and licencing assays. These include the National Institute for Biological Standards and Control in the United Kingdom, respected internationally (36). The WHO can also set up accredited centres to support activities and maintain standards.
The decision to license a COVID-19 vaccine will likely be taken by governments in consultation with manufacturers, advisory bodies and regulators.
Final considerations
Remember, the first COVID-19 vaccines may be in short supply, locked into particular countries, expensive to buy. The whole process needs to be considered from the start to make a low cost, quality vaccine.
We need multiple approaches to making a COVID-19 vaccine, and there are currently >100 in development (37). Having several vaccines is much better than having one. Some vaccines may be more appropriate for use in certain settings e.g. heat stable vaccines. Many vaccines require expensive cold chain refrigeration systems to be distributed. Heat-stable vaccines may be more appropriate for stockpiling and in certain resource limited settings.
If we use relatively novel approaches to vaccinating against COVID-19 we will need to monitor carefully safety, immunogenicity, protection and duration of immunity.
Acknowledgments
The authors would like to thanks Yorgo Modis, Christine Hale and Sophie Palmer for their support in preparing this Blog. The details outlined are our interpretation of what is an incredibly complicated process of making vaccines and openly recognise that others may have their own views on some of the details and the outlined paths. We respect this.
Bibliography
(All links are to open access data)
- https://en.wikipedia.org/wiki/Vaccine
- https://www.cdc.gov/vaccines/vpd/vpd-vac-basics.html
- https://www.vaccines.gov/basics/vaccine_ingredients
- https://www.apm.org.uk/resources/what-is-project-management/
- http://www.euvaccine.eu/vaccines-diseases/vaccines/stages-development
- https://www.who.int/immunization_standards/national_regulatory_authorities/role/en/
- https://www.fda.gov/home
- https://www.ema.europa.eu/en
- https://www.who.int/rhem/prequalification/prequalification_of_medicines/en/
- https://www.gov.uk/guidance/apply-for-a-licence-to-market-a-medicine-in-the-uk
- https://www.oecd.org/chemicalsafety/testing/good-laboratory-practiceglp.htm
- https://www.omicsonline.org/open-access/trends-in-adjuvant-and-vaccine-delivery-systems-mohanty-nn-2332-0877-1000260.php?aid=67725
- https://www.sciencedirect.com/science/article/pii/S0092867420302622
- https://www.ncbi.nlm.nih.gov/pmc/articles/PMC3253344/
- https://en.wikipedia.org/wiki/Protein_production
- https://pichia.com/welcome/
- https://www.youtube.com/watch?v=jx_J_VldcNk
- https://insight.jci.org/articles/view/123158
- https://www.ncbi.nlm.nih.gov/pmc/articles/PMC5609467/
- https://en.wikipedia.org/wiki/Neutralizing_antibody
- https://www.nature.com/articles/227561a0.pdf
- https://www.phgfoundation.org/briefing/rna-vaccines
- https://www.youtube.com/watch?v=8uECsQ441XE
- https://ispe.org/initiatives/regulatory-resources/gmp
- https://www.hillemanlabs.org
- https://www.biopharmainstitute.com/faq/what-is-the-difference-between-single-blind-and-double-blind-clinical-trials
- https://www.ncbi.nlm.nih.gov/pmc/articles/PMC2897268/
- https://acmedsci.ac.uk/policy/policy-projects/controlled-human-infection-models
- https://en.wikipedia.org/wiki/Vaccine_efficacy
- https://www.who.int/bulletin/volumes/91/4/12-113480/en/
- https://www.ema.europa.eu/en/documents/scientific-guideline/draft-guideline-clinical-evaluation-vaccines-revision-1_en.pdf
- https://blog.technavio.com/blog/top-10-vaccine-manufacturers
- https://www.who.int/immunization/programmes_systems/supply_chain/benefits_of_immunization/en/
- https://www.who.int/cholera/vaccines/en/
- https://doi.org/10.1093/infdis/jiaa152
- https://www.nibsc.org
- https://vac-lshtm.shinyapps.io/ncov_vaccine_landscape/
Gordon Dougan FRS is a Professor in the Department of Medicine, University of Cambridge and has spent his career leading research into vaccines, pathogen genomics and disease tracking. His research work has helped to redefine our understanding of how infections spread around the world, a subject of direct relevance to the current Covid-19 epidemic.
Kat Holt is a Professor in the Department of Infectious Diseases, Monash University and the Department of Infection Biology, London School of Hygiene and Tropical Medicine. She is an expert in microbial genomic epidemiology and her research focuses on transmission, evolution and antimicrobial resistance of bacterial pathogens.

One thought on “How to make a new COVID-19 vaccine starting from scratch”
Comments are closed.